Clinical case # 1 (Author: Tom Ferkol, MD)
An eleven-year-old young man with persistent cough.
The patient is an 11-year-old male child who presented to St. Louis Children's Hospital for evaluation of persistent cough.
The term product of an uncomplicated pregnancy and delivery, though labor was induced. The patient's neonatal course was eventful. He developed respiratory distress shortly after birth, and required endotracheal intubation and mechanical ventilator support for approximately one week. He continued to have a supplemental oxygen requirement until ten days of age. Chest radiographs obtained while he was in the Neonatal Intensive Care Unit (NICU) demonstrated situs inversus totalis. He does not have congenital cardiac lesions.
The patient has had several acute otitis media, beginning in early infancy and requiring tympanostomy tube placement. According to his mother, he had transient hearing loss. The patient frequently had problems with otorrhea, particularly after tympanostomy tubes were placed. He has had recurrent nasal symptoms and rhinosinusitis beginning at one month of age. He has not had nasal polyposis. Computerized tomography of the paranasal sinuses performed during early childhood showed mucosal thickening and opacification of the sinuses. He typically has two or three episodes of bacterial sinusitis per year, each requiring oral antibiotic therapy. The patient underwent adenotonsillectomy, but has not required endoscopic sinus surgery.
The patient has had a chronic, productive cough since infancy. He has been diagnosed with recurrent "bronchitis", requiring treatment with oral or intravenous antibiotics. He has been hospitalized three times for the management of his lung disease. He has not had other respiratory symptoms, including wheezing, dyspnea, tachypnea, hemoptysis, or shortness of breath. He has not had apparent exercise intolerance, no nighttime symptoms were noted.
His past medical history is otherwise unremarkable. He has had no known infectious exposures, including pertussis and tuberculosis. His immunizations are current. The patient does not have any allergies. He has not required supplemental oxygen therapy. He seldom performs airway clearance techniques. His current medications include Flovent (fluticasone) metered-dose inhaler, 110 mcg per actuation, 2 puffs administered twice daily; and albuterol meter dose inhaler, 2 puffs administered when symptomatic.
He lives with his parents and three older siblings in a suburban single-family home in Kansas, USA. No one at home is currently ill. The family history is remarkable for his maternal grandmother who underwent lobectomy for chronic obstructive pulmonary disease (COPD). His maternal great grandmother had emphysema, but no cystic fibrosis, recurrent pneumonias, infertility, pancreatitis, retinitis pigmentosa, congenital heart disease, or polycystic kidney disease has been noted in any other family member.
On physical examination, the patient was a pleasant young man in no respiratory distress. His weight was 33.5 kg and height was 145.2 cm. The respiratory rate was 28 breaths/minute. The nasal pharyngeal mucosa was mildly inflamed, but no nasal polyps or discharge was present. The tympanic membranes were gray, but bilateral tympanosclerosis and serous middle ear effusions were evident. The oropharyngeal mucosa was normal. His neck was supple without masses or lymphadenopathy. No stridor was appreciated. The lungs were clear to auscultation with good air exchange. Breath sounds were symmetric. No retractions were noted. Cardiovascular examination revealed a regular rate and normal heart sounds. No murmurs or gallops were present. A right-sided apical impulse was palpated. The abdomen was soft, nontender, and nondistended. Bowel sounds were present. No masses or hepatosplenomegaly was palpated. The extremities were without digital clubbing, cyanosis, or edema.
Pulmonary function studies were obtained today were interpreted as normal, with a forced vital capacity (FVC) and forced expiratory volume in one second (FEV1) of 94% and 88% predicted for age, respectively. There was no bronchodilator response.
Chest radiographs obtained during this visit were interpreted as normal with the exception of situs inversus totalis. There were no focal infiltrates or atelectasis. No pleural effusions were noted. The cardiothymic silhouette size and contour are normal.
High-resolution computerized tomography of the chest was performed which showed clear lung fields without parenchymal nodules or alveolar consolidations. Trachea and major bronchi are normal, but minimal central bronchiectasis was noted involving the lower lobes. Multiple small axillary lymph nodes are observed, but no mediastinal or hilar lymphadenopathy was present. Dextrocardia with right-sided arch and branching of the aorta was noted. In the upper abdomen, the liver was located on the left. A single spleen was seen on the right.
Based on his clinical presentation, he underwent nasal mucosal biopsy, which demonstrated normal ciliary axonemal ultrastructure.
Nasal nitric oxide concentrations were measured and found to be markedly low (10.5 mL/mn).
Blood was collected for genetic testing for primary ciliary dyskinesia-causing mutations in this patient, which revealed two mutant DNAH11alleles (IVS33G>A/L4354H).
Discussion
This young man has primary ciliary dyskinesia (PCD), based on his classic clinical phenotype, reduced nasal nitric oxide levels, and identification of two mutantDNAH11 alleles.
The case also illustrates the limitations of current diagnostic testing. Historically, the diagnosis of PCD required the presence of characteristic clinical phenotype, which include situs inversus totalis with any respiratory disease, unexplained neonatal respiratory distress, persistent nasal rhinitis, daily cough, or bronchiectasis without a defined etiology, and until recently, the identification of ciliary ultrastructural defects using transmission electron microscopy [1]. Recent work defining the genetic basis of PCD has shown that this approach has significant drawbacks and cannot be considered the “gold standard” for diagnosis. Truncation or absence of dynein arms is a common abnormality seen in PCD, but other axonemal changes consistent disease PCD include microtubular transposition, radial spoke with central apparatus abnormalities, and nexin link defects. It is important to note that airway injury or inflammation can also lead to “acquired” ultrastructural defects. Ciliary disorientation was once proposed as a form of PCD, but this finding is a “secondary’ change and should not be used as the basis for diagnosis [1].
As this case illustrates, normal ciliary ultrastructure does not exclude PCD, and is found in approximately 30% of PCD patients, many confirmed by genetic testing. A recent large study showed that 22% of patients with PCD and normal ciliary ultrastructure have disease-causing mutations in DNAH11 [2]. Mutations in other genes, such as CCDC39 or CCDC40, lead to defects involving the inner dynein arm and central apparatus, but only in a small number (5-20%) of ciliary axonemes examined [3,4].
Alternative diagnostic tests have been proposed. Ciliary beat frequency measurements have been advocated as a diagnostic test, but this approach has significant limitations. Airway epithelial inflammation or injury can alter cilia motion, and many forms of PCD have only subtle abnormalities of ciliary beat frequency and waveform. Nasal nitric oxide (NO) measurement is a promising diagnostic approach, since levels in PCD subjects are reproducibly low. Because of overlap in measured nasal nitric oxide levels, cystic fibrosis must be been excluded. Currently in North America, nasal nitric oxide analysis is available only at a few specialized centers.
Genetic testing for PCD is becoming a reality, and commercial laboratories now offer testing for selected disease-causing mutations. The genetic basis of PCD has rapidly emerged, and it is likely that more comprehensive genetic testing will become available, which will serve as a more reliable diagnostic test and allow us to identify individuals who have milder variants and extend the phenotypic spectrum of the disease.
Images
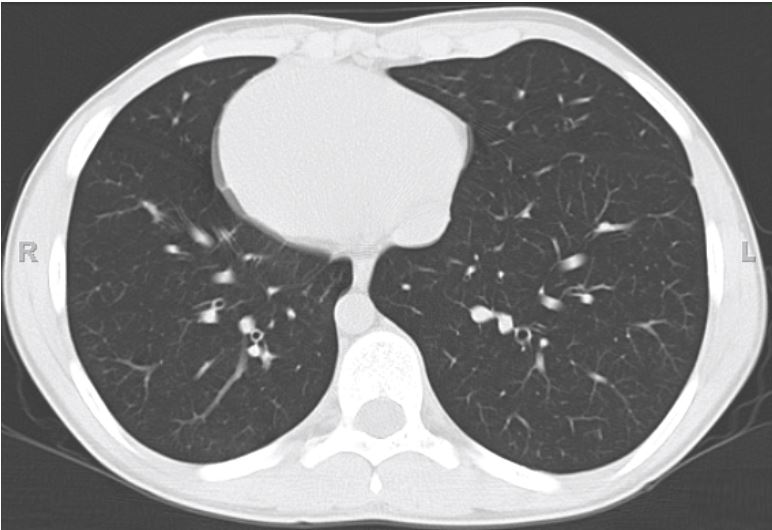
- Computed x-ray tomographic image of the chest that showed dextrocardia with mirror image branching of the thoracic aorta, clear lung fields, and no bronchiectasis or atelectasis. More caudal images revealed left-sided liver and single, right-sided spleen.
References
Ferkol TW, Leigh MW. Ciliopathies: the central role of cilia in a spectrum of pediatric disorders. J Pediatr; 2012;160:366-71
Knowles MR, et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 2012;67:433-441.
Merveille AC, et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat.Genet. 2012;43:72-78.
Becker-Heck A, et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat.Genet. 2011;43:79-84.